Należy odróżniać tę chorobę od dziedziczonego w sposób recesywny wczesnego zwyrodnienia siatkówki (early retinal degeneration – erd), częściej opisywanego u tej rasy. W tym typie PRA ślepota nocna pojawia się w szóstym tygodniu życia, a w wieku 12-18 miesięcy psy są całkowicie ślepe. Zmiany morfologiczne wewnętrznych i zewnętrznych warstw pręcików są obecne w piątym tygodniu życia, a zarówno zakończenia synaptyczne pręcików, jak i czopków rozwijają się nieprawidłowo. Wykryto odpowiedzialną za chorobę mutację genu (STK38L) i opracowano test DNA.
Czwarty typ PRA, opisywany jako dysplazja fotoreceptorów (photoreceptor dysplasia – pd), stwierdzano u sznaucerów miniaturowych. Choroba dziedziczy się w sposób autosomalny recesywny, a jej niezwykłą cechą jest to, że pomimo nieprawidłowego rozwoju czopków i pręcików od 24. dnia życia, klasyczne zmiany w badaniu oftalmoskopowym u chorych psów widoczne są nie wcześniej niż po ukończeniu drugiego roku. Ostatnio uważa się, że u tej rasy występuje również drugi (a możliwe, że także trzeci) typ PRA. Inny rodzaj dysplazji fotoreceptorów opisano u owczarków belgijskich, ale w ich przypadku do całkowitej ślepoty dochodzi w wieku ośmiu tygodni. Pofałdowanie siatkówki widoczne jest od jedenastego tygodnia życia, a fałdy stopniowo zastępowane są przez obszary zwyrodnienia, charakteryzujące się wzmocnieniem odblasku i centralną pigmentacją.
Pomimo różnic w fenotypie i w szybkości postępowania objawów klinicznych choroby, genetycznie podobny typ pojawiającego się w późniejszym okresie PRA – postępujące zwyrodnienie czopków i pręcików (progressive rod-cone degeneration – prcd) – opisano u dziewięciu ras psów. Choroba dziedziczy się w sposób recesywny, a różnice w objawach klinicznych wynikają z obecności różnych mutacji w locus prcd lub tej samej mutacji, ale o ekspresji zmienionej przez czynniki genetyczne. Rasy predysponowane to amerykański i angielski cocker spaniel, australijski pies pasterski, chesapeake bay retriever, labrador retriever, pudel miniaturowy i toy, nova scotia duck tolling retriever oraz portugalski pies dowodny.
U obu odmian pudli rozpoznanie na podstawie badania oftalmoskopowego jest możliwe między trzecim a piątym rokiem życia, a ślepota pojawia się w wieku 5-7 lat. Widoczne są szarawe odbarwienia części jasnej siatkówki, następnie dochodzi do osłabienia naczyń krwionośnych i wzmocnienia odblasku, początkowo w strefach środkowo-obwodowej i obwodowej. Często powstaje wtórna zaćma. Fotoreceptory rozwijają się prawidłowo aż do czternastego tygodnia życia, gdy staje się widoczna dezorganizacja ich zewnętrznych segmentów.
U cocker spanieli angielskich prcd zwykle ujawnia się klinicznie w późniejszym okresie – opisywano przedział wiekowy między trzecim a ósmym rokiem życia. Wygląd dna oka może być różny – smugi wzmocnionego odblasku bywają widoczne centralnie po jednej stronie tarczy nerwu wzrokowego, podczas gdy inne obszary siatkówki wyglądają prawidłowo. Różnice widoczne w badaniu oftalmoskopowym znajdują odzwierciedlenie w postaci zmian histologicznych, które w tej samej siatkówce mogą być jednocześnie zaawansowane i łagodne. Taka niejednorodność zmian jest typowa u omawianej rasy. Tempo rozwoju choroby u pudli jest też zwykle wolniejsze. Również u tej rasy często powstaje wtórna zaćma.
U amerykańskich cocker spanieli z prcd utrata widzenia nocnego następuje pomiędzy trzecim a piątym rokiem życia, a w ciągu 24 miesięcy od wystąpienia tego objawu psy zwykle całkowicie ślepną. Zmiany w badaniu oftalmoskopowym są widoczne już w wieku 2,5 roku, ale nieregularność zwyrodnienia, obserwowana często u cocker spanieli angielskich, nie jest typową cechą u ich amerykańskich kuzynów. Zmiany na dnie oka są podobne do tych obserwowanych u pudli.
U labradorów utrata nocnego widzenia nie występuje do 4.-6. roku życia, a poważne upośledzenie wzroku bywa obserwowane dopiero w wieku 7 lub 8 lat. Zmiany odblasku są widoczne w części środkowo-obwodowej i obwodowej dna oka. Często towarzyszą im brązowawe smugi po obu stronach tarczy nerwu wzrokowego. Następnie dochodzi do wzmocnienia odblasku i osłabienia naczyń krwionośnych, zmiany w centralnej części dna oka mogą jednak nie być wyraźnie zauważalne. U portugalskich psów dowodnych obraz kliniczny jest podobny jak u labradorów, a wczesne rozpoznanie na podstawie badania oftalmoskopowego jest możliwe między trzecim a szóstym rokiem życia.
Dziedziczone w sposób autosomalny recesywny, sprzężone z chromosomem X zwyrodnienie siatkówki (x-linked, autosomal recessive hereditary retinal degeneration – XLPRA), występujące u husky syberyjskich i samojedów, objawia się w wieku 2-4 lat, a choroba klinicznie przypomina prcd spotykane u pudli. Opisano mutację genu RPGR i opracowano test DNA.
Dystrofie czopków i pręcików o wczesnym początku (Cone-Rod Dystrophies – CORD/crd) opisywano u amerykańskich pitbull terrierów, jamników gładkowłosych, szorstkowłosych i miniaturowych długowłosych. W badaniu elektroretinograficznym amplituda impulsów swoistych dla czopków ulega znacznemu obniżeniu w wieku 5-6 tygodni, podczas gdy sygnały emitowane przez pręciki są prawidłowe. U jamników zidentyfikowano mutację genu RPGRIP1 (retinitis pigmentosa GTPase regulator-interacting protein 1) i opracowano test do jej wykrywania. U springer spanieli angielskich wykryto również mutację CORD1 jako drugi typ PRA u tej rasy. Pojawiająca się w późniejszym okresie życia dystrofia czopków i pręcików (crd3) została przypisana delecji eksonów 15 i 16 genu ADAM9 u rasy glen of imaal terrier i obecnie dostępny jest test DNA (5).
Pozostałe rodzaje PRA, z wyjątkiem jednego, określane są po prostu terminem „postępujący zanik siatkówki” (PRA). Przeprowadzono dokładne badania histologiczne i elektrofizjologiczne u akita, angielskich springer spanieli, papillonów, spanieli tybetańskich i terierów tybetańskich. U powyższych ras wiek, w którym pojawia się choroba, jest różny. U papillonów utrata widzenia nocnego i zmiany w badaniu oftalmoskopowym pojawiają się zwykle dopiero w średnim wieku. U tej rasy choroba przebiega podobnie do prcd u pudli, ale nie występuje wtórna zaćma. U spanieli tybetańskich drobne zmiany na dnie oka i ślepota nocna pojawiają się pomiędzy trzecim a piątym rokiem życia. Jednak u terierów tybetańskich utrata widzenia nocnego występuje w wieku 9-10 miesięcy, a całkowita ślepota często przed trzecim rokiem życia. Lekarze klinicyści powinni pamiętać, że u tej rasy obserwuje się trzy rodzaje PRA, łącznie z opisanym niedawno PRA3, powodującym wystąpienie choroby pomiędzy czwartym a siódmym rokiem życia, oraz PRA pojawiającym się w dużo późniejszym okresie życia. Dodatkowe komplikacje wynikają z faktu, że zwyrodnienie siatkówki spotyka się również w przebiegu lipofuscynozy ceroidowej neuronów, która u tej rasy jest dziedziczna.
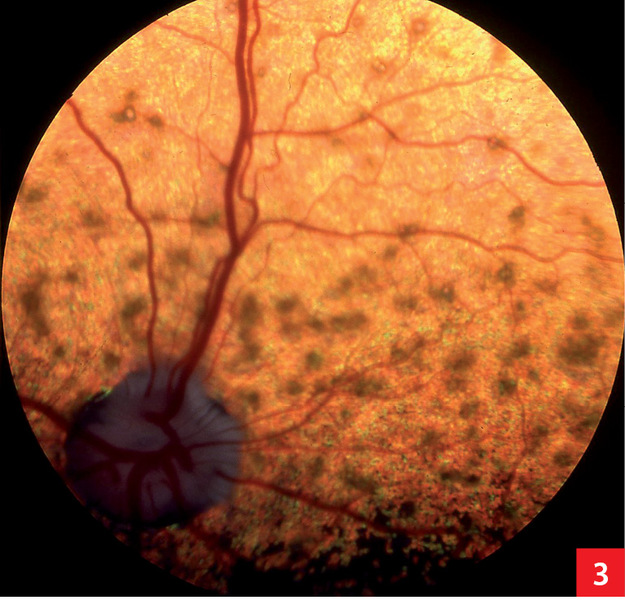
Objawem zwiastunowym PRA u akita jest ślepota nocna pojawiająca się między pierwszym a trzecim rokiem życia. Opisano dwa wzorce wczesnych zmian w badaniu dna oka. Mogą one przybierać postać pasm wzmożonego odblasku rozciągających się od części centralnej na obrzeża lub wzmocnienia odblasku w obwodowej części błony odblaskowej. Najczęstsza forma, z uogólnionym wzmocnieniem odblasku i osłabieniem unaczynienia, prowadzi do całkowitej ślepoty między trzecim a piątym rokiem życia.
U springer spanieli angielskich etiologia choroby nie jest do końca jasna, ponieważ występują znaczne różnice wieku, w którym pojawiają się objawy kliniczne, a wczesne objawy choroby różnią się od tych typowych. Początkowo spotyka się niewielkie zmiany zabarwienia siatkówki, zlokalizowane obwodowo na połączeniu części jasnej i ciemnej dna oka, a następnie pojawia się poziome pasmo zwyrodnienia, powoli przesuwające się w kierunku centralnym. Niekiedy widać uogólnione wzmożenie odblasku i osłabienie unaczynienia. Często tworzy się wtórna zaćma. Nie zidentyfikowano rodzaju mutacji i nie opracowano testu DNA – niestety stwierdzona ostatnio u omawianej rasy mutacja CORD1 nie powoduje tej choroby.
Ponieważ wzrasta świadomość na temat dziedzicznego zwyrodnienia siatkówki u psów rasowych i rozwinęły się badania genetyczne, PRA o późnym początku (late onset PRA – LOPRA) rozpoznaje się obecnie u wielu ras. Dostępny jest test DNA, chociaż szczegółowe badania dotyczące swoistej mutacji nie zostały jeszcze opublikowane. Do momentu opracowania niniejszej publikacji LOPRA stwierdzano u seterów szkockich, irlandzkich i angielskich oraz u terierów tybetańskich, należy się jednak spodziewać, że w przyszłości choroba zostanie wykryta również u innych ras. Istotą choroby jest zwyrodnienie czopków i pręcików i prawdopodobnie przez pomyłkę otrzymała ona nazwę rcd4. Zwykle stwierdza się ją u psów w wieku 8-10 lat i podobnie jak inne zwyrodnienia fotoreceptorów, charakteryzuje się początkowo utratą widzenia nocnego. Dlatego u seterów irlandzkich wyodrębniono dwa typy dziedzicznej choroby siatkówki prowadzącej do ślepoty, o wczesnym i późnym początku. Możliwy jest także trzeci rodzaj zwyrodnienia czopków i pręcików u tej rasy, objawiający się w pośrednim wieku między rcd1 i rcd4.
Wyraźny wyjątek wśród PRA stanowi dziedziczone recesywnie zwyrodnienie czopków (cone degeneration – CD), opisywane u alaskan malamutów, odpowiedzialne za wrodzoną ślepotę dzienną (hemeralopię). Objawy są widoczne u 8-10-tygodniowych szczeniąt, nigdy jednak nie stwierdza się zmian w badaniu oftalmoskopowym. Odruchy źreniczne są prawidłowe i nie dochodzi do utraty widzenia nocnego. Czopki w momencie narodzin są prawidłowe, ale u niektórych psów w wieku siedmiu tygodni widać zmiany w ich morfologii. W wieku sześciu miesięcy wszystkie czopki są zmienione, a w stadium końcowym, w wieku czterech lat, siatkówka składa się już z samych pręcików. Za przyczynę tego rodzaju PRA uważa się selektywny brak ekspresji β3-transducyny z powodu delecji całego genu CNGB3.
Dystrofia nabłonka barwnikowego siatkówki (Retinal Pigment Epithelial Dystrophy – RPED)
Choroba ta, początkowo uważana za pierwotne zwyrodnienie fotoreceptorów, rozwija się wtórnie do nieprawidłowej aktywności komórek nabłonka barwnikowego. Jedną z najważniejszych funkcji tego nabłonka jest degradacja zużytych segmentów zewnętrznych fotoreceptorów (photoreceptor outer segments – POS). Barwnik wzrokowy znajduje się w POS i podczas procesu fototransdukcji następuje szybka wymiana tego materiału. Odbudowa zewnętrznego fragmentu pręcika trwa około dziesięciu dni, a podstawowy budulec dostarczany jest przez komórki nabłonka barwnikowego i pochodzi z przetwarzanych przez nie zużytych POS. Niesprawne komórki RPE nie są w stanie wystarczająco szybko degradować zużytych segmentów pręcików ani efektywnie uczestniczyć w ich wytwarzaniu. W ich cytoplazmie gromadzą się duże ilości sfagocytowanego materiału pochodzącego z POS i nabłonek barwnikowy przestaje pełnić funkcję wspomagającą względem siatkówki nerwowej. Oba rodzaje fotoreceptorów (czopki i pręciki) ulegają zwyrodnieniu i dochodzi do upośledzenia widzenia.
RPED stwierdzano u border collie, owczarków szkockich collie krótko- i długowłosych, owczarków szetlandzkich, cocker spanieli angielskich, springer spanieli angielskich, briardów i polskich owczarków nizinnych. Mechanizm dziedziczenia pozostaje nieznany, na szczęście jednak częstotliwość występowania choroby na świecie szybko się zmniejsza.